Massively parallel sequencing (MPS)–based single nucleotide polymorphism (SNP) profiling is being increasingly used in forensic DNA testing, yet the informative value of common pre-sequencing DNA metrics to predict SNP profile completeness remains unclear. In this study, 500 anonymized skeletal samples submitted for forensic genome sequencing were analyzed to assess the relationships between bone type, DNA quantitation metrics, and SNP call rate (the latter used as a measure of profile completeness). Human-specific DNA quantity was measured using short and long autosomal quantitative PCR targets, and total DNA content was assessed fluorometrically.
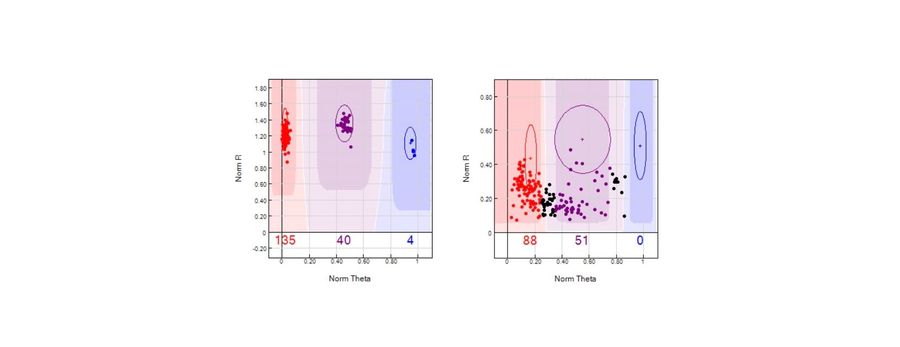
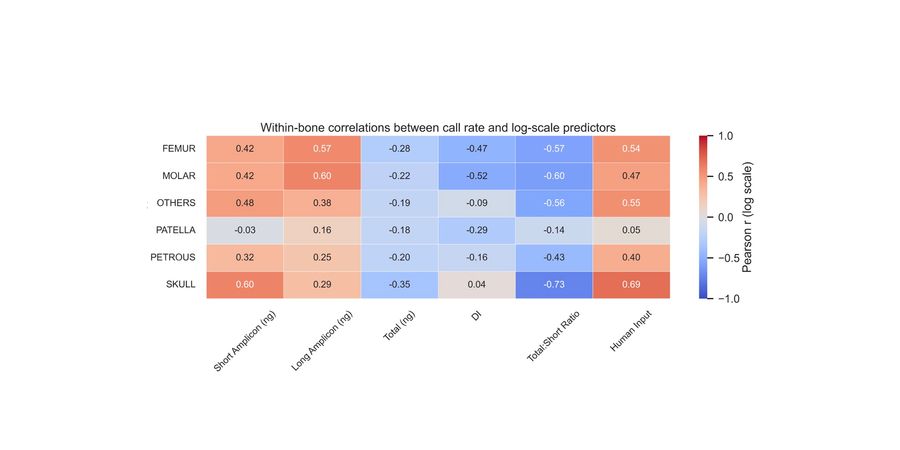
Of the 500 samples, 399 met a minimum human DNA threshold (short autosomal target ≥0.005ng/µl) and were sequenced. Among these samples, SNP call rates ranged from 8% to 91%, with 95.7% achieving call rates greater than 50%. Call rate was most strongly associated with features reflecting the ratio between endogenous human DNA and total DNA, which includes exogenous background DNA. The total:short DNA ratio (log₁₀ r = −0.48) and estimated human DNA input into library preparation (log₁₀ r = 0.41) showed the strongest correlations, while degradation index showed only a modest association. Bone type influenced the likelihood that samples progressed to sequencing, but among sequenced samples, call rate distributions were similar across major bone type categories. Machine-learning models achieved moderate predictive performance (best validation R² = 0.47), indicating that current quantification metrics are correlated with, but not sufficient to reliably predict, SNP profile completeness.
Overall, these results demonstrate that MPS-based SNP profiling of UHRs is highly effective and support forensic genetic genealogy as the preferred approach for generating actionable genetic data for identification of human remains. However, due to varying degrees of DNA quality and quantity in bones samples, the common pre-sequencing metrics, while providing some actionable workflow decisions in this study, are insufficient predictors for the range of samples encountered in unidentified human remains.